

Электрофильные реакции
В основе Э. р. лежит
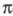 -электронодонорная
способность олефинов, ацетиленов и ароматич. углеводородов по отношению
к электрофилам, а также возможность передачи гетероатомами и простыми связями
С — С и С — Н своих электронных пар.
-электронодонорная
способность олефинов, ацетиленов и ароматич. углеводородов по отношению
к электрофилам, а также возможность передачи гетероатомами и простыми связями
С — С и С — Н своих электронных пар.
К р-циям электроф. замещения в алифатич. ряду относятся р-ции обмена металлов (гл. обр. ртути) в металлоорг. соед. на другой металл, водород или галоген; водородно-дейтериевый обмен; р-ции изомеризации и др.
Возможны три механизма р-ций: мономолекулярный SE1 и бимолекулярные SE2и SE i:
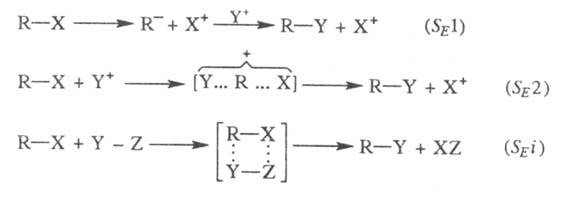
В этих р-циях Y+ - электрофил,
Х+ - электронодефицитная уходящая группа, наз. электрофугом
(от лат. fugio -убегаю).
При мономол. процессе SE1происходит
ионизация субстрата с образованием карбаниона (эта стадия обычно определяет
скорость р-ции), а затем быстрая стадия связывания карбаниона с электрофилом.
В ходе р-ций по этому механизму происходит, как правило, рацемизация, хотя
в нек-рых случаях возможно сохранение или даже обращение конфигурации.
В р-циях, протекающих по механизму SE2,
возможна атака электрофила со стороны уходящей группы (при этом конфигурация
субстрата сохраняется) и с противоположной стороны (конфигурация обращается).
В первом случае уходящая группа может отделяться одновременно с образованием
новой связи (механизм SEi);
в этом случае также наблюдается
сохранение конфигурации.
Механизм электроф. замещения зависит от
природы, субстрата и р-рителя, как и в случае нуклеоф. замещения. Повышение
полярности р-рителя увеличивает возможность ионизации в р-циях, протекающих
по механизму SE1, а также ускоряет р-ции типа SE2,
тогда как на р-ции типа SEi влияет гораздо меньше.
Большое влияние оказывает природа электрофуга: в случае карбкатионных электрофуюв
наблюдается механизм SE1, тогда как для металлсодержащих
- SE2 или SE i.
Заместители в субстрате, обладающие отрицат.
индуктивным и мезомерным эффектами, ускоряют р-ции типа SE1.
Р-ции типа SE2также ускоряются при наличии заместителей,
обладающих отрицат. индуктивным эффектом, и замедляются при наличии заместителей
с положит. индуктивным эффектом. Так, относит. скорость изотопного замещения
ртути CH3HgX + Hg*X2 CH3Hg*X
+ HgX2 возрастает в 8 раз при переходе от Х = Вг к Х = 1 и в
240 000 раз при переходе от X = ОСОСН3 к X = NО2.
CH3Hg*X
+ HgX2 возрастает в 8 раз при переходе от Х = Вг к Х = 1 и в
240 000 раз при переходе от X = ОСОСН3 к X = NО2.
В нек-рых случаях электроф. замещение
может сопровождаться перегруппировками углеродного скелета молекулы. Напр.,
бензиновые эфиры при действии литийорг. агентов изомеризуются в спирты
(перегруппировка Виттига):
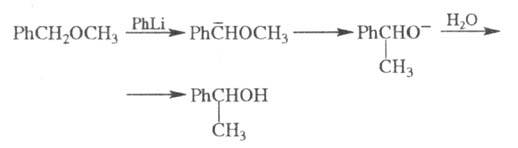
Для аммониевых солей характерна
Стивенса
перегруппировка. Описаны также электроф. перегруппировки Клайзена,
Фриса и др.
Один из важнейших типов р-ций в орг. химии
- электроф. замещение в ряду ароматич. со ед.; последние благодаря наличию -системы
легко создают центры с повышенной электронной плотностью. К таким р-циям
относят нитрование, нитрозирование, ацилирование по Фриделю-Крафтсу и др.
-системы
легко создают центры с повышенной электронной плотностью. К таким р-циям
относят нитрование, нитрозирование, ацилирование по Фриделю-Крафтсу и др.
В начальной стадии электрофил Y+
образует с ароматич. субстратом промежут. комплекс (I). Обычно считают,
что для ароматич. соед., активированных электронодонорными заместителями,
структура (I) соответствует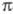 -комплексу,
в к-ром Y* расположен над плоскостью кольца (впервые концепция
-комплексу,
в к-ром Y* расположен над плоскостью кольца (впервые концепция -комплексов в Э. р. была выдвинута М. Дьюаром в 1946):
-комплексов в Э. р. была выдвинута М. Дьюаром в 1946):
Образование -комплекса,
как правило, характеризуется очень высокими скоростями (до 1010
с-1). Лимитирующая стадия - формирование циклогексадиенильного
катиона (П), т. наз.
-комплекса,
как правило, характеризуется очень высокими скоростями (до 1010
с-1). Лимитирующая стадия - формирование циклогексадиенильного
катиона (П), т. наз.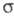 -комплекса
(комплекса Уэланда, или аренониевого иона), либо, реже, распад II через
промежуточный
-комплекса
(комплекса Уэланда, или аренониевого иона), либо, реже, распад II через
промежуточный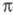 -комплекс
III (см. также рис.). Образование
-комплекс
III (см. также рис.). Образование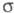 -комплексов
доказано в нек-рых газофазных р-циях электроф. замещения с помощью радиохим.
методов, масс-спектрометрии и ион-циклотронного резонанса; в условиях высокого
вакуума
-комплексов
доказано в нек-рых газофазных р-циях электроф. замещения с помощью радиохим.
методов, масс-спектрометрии и ион-циклотронного резонанса; в условиях высокого
вакуума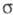 -комплексы
м. б. достаточно устойчивыми. Из р-ров препаративно выделены соли ряда
катионов II. При использовании высокореакционноспособных реагентов лимитирующей
стадией может стать стадия образования
-комплексы
м. б. достаточно устойчивыми. Из р-ров препаративно выделены соли ряда
катионов II. При использовании высокореакционноспособных реагентов лимитирующей
стадией может стать стадия образования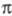 -комплекса.
-комплекса.
В р-циях электроф. замещения монозамещенных
производных бензола новая группа вступает в орто-, мета-
или пара-положение;
при этом заместители либо облегчают, либо затрудняют протекание р-ции.
По правилам ориентации заместители, проявляющие положит. индуктивный эффект
(+I) и положит. мезомерный эффект (+М), активируют ароматич.
ядро и являются орто- или пара-ориентантами. Заместители,
проявляющие -I и +М-эффекты, также орто-
или пара-ориентанты;
однако, в случае когда индуктивный эффект больше мезомерного (напр., у
Hal), ароматич. ядро пассивируется, если же индуктивный эффект меньше мезомерного
(OR, SR, NR2 и др.), - ядро активируется. Заместители, проявляющие
-I и -М-эффекты (CN, NO2), пассивируют ядро и
являются мета-ориентантами (см. табл.). По иной классификации орто-
и пара-ориентанты относят к заместителям I рода, а мета-ориентанты
- к заместителям II рода. Причина разл. реакц. способности орто-, мета-
и
пара-положений - изменение распределения электронной плотности в
кольце под влиянием заместителей.
КЛАССИФИКАЦИЯ ЗАМЕСТИТЕЛЕЙ В АРОМАТИЧЕСКОМ
ЯДРЕ В ЭЛЕКТРОФИЛЬНЫХ РЕАКЦИЯХ
|
Заместители
|
Тип электронного
эффекта
|
Влияние на скорость
реакции
|
Тип ориентации
|
|
Ал килы
|
+I,M
= 0
|
Ускоряют
|
орто-, пара-
|
|
O-
, OR, NRR', С6Н5, СН = СН2
|
+М>-I
|
Ускоряют
|
орто-, пара-
|
|
Hal, (C6H5)2O+
|
-I>+M
|
Замедляют
|
орто-, пара-
|
|
CHal3,
(CH3)3N+
|
-I,
М
= 0
|
Замедляют
|
мета-, пара-
|
|
NO2,
CN, CHO, COR, COOR, COHal
|
-1,-М
|
Замедляют
|
мета-
|
|
CH2Cl,
CH2NO2, CH2OH
|
+I,-I,
М
= 0
|
Замедляют
|
орто-,мета-,
пара-
|
Соотношение орто-, мета-
и пара-изомеров
в продуктах р-ции зависит от природы электроф. агента. Так, при хлорировании
толуола образуется до 75% opтo-производного, а при меркурировании
- до 80% пара-замещенного продукта.
Для количеств, характеристики ориентирующего
влияния заместителей используют факторы парциальной скорости (ФПС), т.
е. скорости замещения в определенное положение ароматич. ядра относительно
скорости замещения в одно положение бензола. ФПС выражают через константы
скорости по всем положениям для замещенного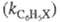 и незамещенного
и незамещенного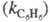 продуктов и долю каждого изомера (z) в процентах:
продуктов и долю каждого изомера (z) в процентах: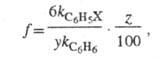
где у - число эквивалентных положений
(для орто- и меma-изомеров у = 2, для пара-изомера
у
= 1).
Для эксперим. определения ФПС используют
конкурирующее замещение в смеси С6Н6 и С6Н5Х
при действии к.-л. электрофила. Возникающая смесь продуктов анализируется
с определением орто-, мета- и пара-замещенных типа YC6H4X,
а также производного бензола YC6H5.
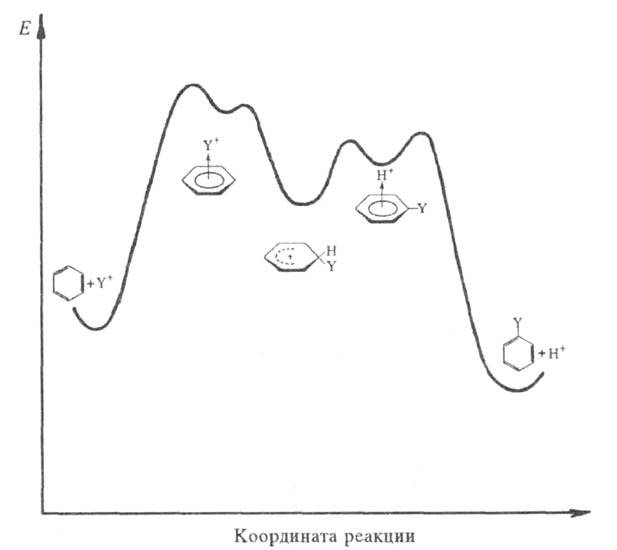
Схематич. изображение изменения энергии системы в р-циях электроф. замещения в ароматич. ядре.
Электроф. замещение в ароматич. ряду подчиняется
ур-нию Гаммета (см. Корреляционные соотношения). Однако последнее
неприменимо для opmo-положений, поскольку не учитывает пространств.
факторы. Используя электроф. константы заместителей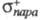 и
и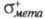 , можно
вычислить парциальные скорости образования пара- и мета-изомеров:
, можно
вычислить парциальные скорости образования пара- и мета-изомеров:
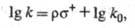
ще k и k0- константы
скорости образования пара(мета)-изомера монозамещенного бензола
и незамещенного бензола соответственно.
Для характеристики селективности электрофила
по отношению к субстрату используют фактор селективности (Sf),
к-рый
определяют по ур-нию:
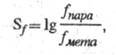
где fпара и fмета
- ФПС для пара- и мета-замещения ароматич. соед. В р-циях
галогенирования и ацилирования по Фриделю-Крафтсу проявляется высокая селективность
злектрофила по отношению к субстрату, протонирование и нитрование занимает
промежут. положение, а алкилирование по Фриделю-Крафгсу малоселективно.
Весьма распространены р-ции электроф.
присоединения к двойной и тройной связи С — С (АdЕ-реакции). Как
правило, эти процессы рассматривают как двухступенчатые, на лимитирующей
стадии к-рых образуется катионоидный интермедиат (ионный путь а):
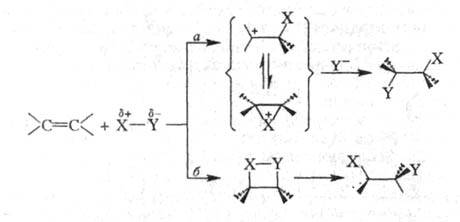
В нек-рых случаях (напр., при взаимод. олефинов с BR3, HHal и др.) присоединение может осуществляться по мол. механизму (путь б).
Во мн. случаях р-ции AdE идут стереоселективно (напр., присоединение галогенов приводит к анти-аддуктам). Электроф. присоединение полярных молекул к алкенам и алкинам протекает по Марковникова правилу и Зайцева-Вагнера правилу.
Лит.: Реакционная способность и пути реакции, пер. с англ., М., 1977; Смит В. А., "Изв. Сиб. отд. АН СССР", сер. хим. н., 1980, в. 3, № 7, с. 128-38; Кери Ф., Сандберг Р., Углубленный курс органической химии, пер. с англ., М., 1981; Общая органическая химия, пер. с англ., т. 1, М., 1981, с. 314-^55; Минкин В. И., Снмкнн Б. Я., Миняев P.M., Квантовая химия органических соединений. Механизмы реакций, М., 1986; Марч Дж., Органическая химия, пер. с англ., т. 2, М., 1987, с. 304-502; Агрономов А. Е., Избранные главы органической химии, 2 изд., М., 1990; Шабаров Ю. С., Органическая химия, т. 1-2, М., 1994.
М. Е. Клецкий.